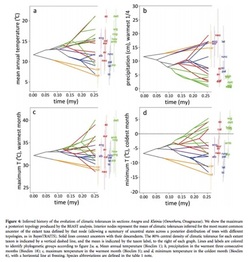
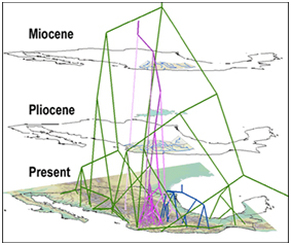
In their 2009 paper “Climate, Niche Evolution, and Diversification of the“Bird-Cage” Evening Primroses”, Evans et al. integrate climatic niche models and dated phylogenies to characterize the evolution of climatic niches in several taxonomic groups of the evening primrose. To do this, MAXENT was used to predict species occupancy of climatic niches. From this they quantified climatic disparity among taxa which was then used as a proxy for how niches have evolved. Using phylogenetic trees and niche differences, they inferred the evolutionary history of climatic tolerances for the primrose taxa in the southwestern US and California. They inferred that the speciation has occurred in response to temperature variation in past climates across this region.
This is an exciting application that combines phylogenetic, species distribution and environmental data. However, there are some assumptions at the core of environmental niche modeling that should be highlighted when considered this type of analysis. First of all, just because an organism is found in an area does not mean that this is its optimal niche. A population may be in an area that was previously its optimal niche or was the niche it evolved
into, but, due to changing climate, may now be in a suboptimal habitat, stuck as a relict. This is often the case for source and/or sink populations and may most likely occur for long-lived species like corals or large trees. Another issue is that the variables you choose for your environmental niche model may not be the key factors driving
presence or absence of a population. Say for example you chose temperature and precipitation variables to build you niche models, but actually, soil or species interactions are more important. If niche evolution models are of
interest, this could be very important because you are assuming the strongest correlated niche variables are drivers of speciation. Finally, when using environmental data, much of it is available in very course scales and may not
really represent on-the-ground ecology or natural selection. For example Bioclim is often available in 30” grids (1 km2 at the equator). Within each grid there may be a tremendous amount of temperature and moisture variability due to micro-topography, slope direction, vegetation, soil type, etc. It may be worth spending time on the ground looking in detail at the organism’s actual niche to see if any micro-patterns emerge.
Evans et al. do clearly mention these issues and try to control for them. The issues are universal problems for niche modeling and difficult to avoid. Several ways to confirm that the plant species they examined are truly in optimal niches would be to out-plant one species into another species’s niche space. This could be done in controlled greenhouse experiments and in the field. If the plants survive the switch and are able to reproduce, the original niche spaces may not be optimal. One caveat is that species may live in a place because of their ability to tolerate extreme climate events which do not happen very often. Another interesting thing to do would be to look at gene expression during the maximum and minimum temperature events for different species. This would tie environmental variables with genetic expression. I enjoyed this paper and recommend it to others.
This is an exciting application that combines phylogenetic, species distribution and environmental data. However, there are some assumptions at the core of environmental niche modeling that should be highlighted when considered this type of analysis. First of all, just because an organism is found in an area does not mean that this is its optimal niche. A population may be in an area that was previously its optimal niche or was the niche it evolved
into, but, due to changing climate, may now be in a suboptimal habitat, stuck as a relict. This is often the case for source and/or sink populations and may most likely occur for long-lived species like corals or large trees. Another issue is that the variables you choose for your environmental niche model may not be the key factors driving
presence or absence of a population. Say for example you chose temperature and precipitation variables to build you niche models, but actually, soil or species interactions are more important. If niche evolution models are of
interest, this could be very important because you are assuming the strongest correlated niche variables are drivers of speciation. Finally, when using environmental data, much of it is available in very course scales and may not
really represent on-the-ground ecology or natural selection. For example Bioclim is often available in 30” grids (1 km2 at the equator). Within each grid there may be a tremendous amount of temperature and moisture variability due to micro-topography, slope direction, vegetation, soil type, etc. It may be worth spending time on the ground looking in detail at the organism’s actual niche to see if any micro-patterns emerge.
Evans et al. do clearly mention these issues and try to control for them. The issues are universal problems for niche modeling and difficult to avoid. Several ways to confirm that the plant species they examined are truly in optimal niches would be to out-plant one species into another species’s niche space. This could be done in controlled greenhouse experiments and in the field. If the plants survive the switch and are able to reproduce, the original niche spaces may not be optimal. One caveat is that species may live in a place because of their ability to tolerate extreme climate events which do not happen very often. Another interesting thing to do would be to look at gene expression during the maximum and minimum temperature events for different species. This would tie environmental variables with genetic expression. I enjoyed this paper and recommend it to others.


 RSS Feed
RSS Feed
