The lab has been funded as part of a large grant (led by Rosie Gillespie) to examine the origins of Hawaiian biodiversity. Here's a link to the announcement on the ESPM web page.
|
In their 2009 paper “Climate, Niche Evolution, and Diversification of the“Bird-Cage” Evening Primroses”, Evans et al. integrate climatic niche models and dated phylogenies to characterize the evolution of climatic niches in several taxonomic groups of the evening primrose. To do this, MAXENT was used to predict species occupancy of climatic niches. From this they quantified climatic disparity among taxa which was then used as a proxy for how niches have evolved. Using phylogenetic trees and niche differences, they inferred the evolutionary history of climatic tolerances for the primrose taxa in the southwestern US and California. They inferred that the speciation has occurred in response to temperature variation in past climates across this region.
This is an exciting application that combines phylogenetic, species distribution and environmental data. However, there are some assumptions at the core of environmental niche modeling that should be highlighted when considered this type of analysis. First of all, just because an organism is found in an area does not mean that this is its optimal niche. A population may be in an area that was previously its optimal niche or was the niche it evolved into, but, due to changing climate, may now be in a suboptimal habitat, stuck as a relict. This is often the case for source and/or sink populations and may most likely occur for long-lived species like corals or large trees. Another issue is that the variables you choose for your environmental niche model may not be the key factors driving presence or absence of a population. Say for example you chose temperature and precipitation variables to build you niche models, but actually, soil or species interactions are more important. If niche evolution models are of interest, this could be very important because you are assuming the strongest correlated niche variables are drivers of speciation. Finally, when using environmental data, much of it is available in very course scales and may not really represent on-the-ground ecology or natural selection. For example Bioclim is often available in 30” grids (1 km2 at the equator). Within each grid there may be a tremendous amount of temperature and moisture variability due to micro-topography, slope direction, vegetation, soil type, etc. It may be worth spending time on the ground looking in detail at the organism’s actual niche to see if any micro-patterns emerge. Evans et al. do clearly mention these issues and try to control for them. The issues are universal problems for niche modeling and difficult to avoid. Several ways to confirm that the plant species they examined are truly in optimal niches would be to out-plant one species into another species’s niche space. This could be done in controlled greenhouse experiments and in the field. If the plants survive the switch and are able to reproduce, the original niche spaces may not be optimal. One caveat is that species may live in a place because of their ability to tolerate extreme climate events which do not happen very often. Another interesting thing to do would be to look at gene expression during the maximum and minimum temperature events for different species. This would tie environmental variables with genetic expression. I enjoyed this paper and recommend it to others. I was talking with Patrick earlier this week about the effects of large data sets, such as from so-called NextGen sequencing (I dislike that term), on phylogenetic and population genetic methods of analysis. It used to be that the analytical bottleneck was in obtaining sequence data, but that has shifted so that now large amounts of sequence data can be obtained quickly but we are limited by computational power after we have them. I recently read a paper (Tamura et al. 2012, in PNAS) that addressed this problem by bucking the trend for building more and more statistical complexity into Maximum Likelihood and Bayesian analytical packages, and instead introduced a quick and fairly simple method to analyze large phylogenetic datasets with extensive evolutionary rate variation along the branches. The method is implemented in RelTime.
In phylogenetics we try to determine two things, the relationships between taxa and the timing of their divergence, either in relative or absolute time. The most common way to date divergence times is to calibrate a molecular phylogeny using the fossil record. Some form of a molecular clock is then assumed to extrapolate rates of evolution and divergence times to uncalibrated parts of the tree. This extrapolation requires assumptions about if and how rates of sequence evolution vary along different branches of the tree. Phylogenetics programs like BEAST require the user to specify, a priori, a statistical distribution from which to model evolutionary rate heterogeneity among tree branches. This method has been largely successful, but is limited computationally because the complexity increases exponentially as the number of taxa in the tree increases. Thus, the challenge with large datasets is that it can take weeks for even powerful computers to complete each step of the analysis. RelTime attempts to circumvent this computational limitation by calculating relative divergence times rather than placing them in the context of absolute time. To me, it looks similar to Neighbor Joining methods used for creating tree topology, except that the method assumes a bifurcating topology with branch lengths, then uses distance calculations as it allows for rate heterogeneity along any branches of the tree. RelTime computes branch-specific relative rates from the tree by averaging branch lengths for each pair of descendants at each node. Check out the paper if you want to dig into the math. The result is a tree with relative divergence times, which then can be converted to absolute divergence times using only a few fossil calibration points in a post hoc application. I assume geographic calibrations such as island age could just as easily be used to calibrate the tree. The biggest advantage of this method is that it is very fast – the authors claim it is 1000 times faster than MCMCTree, which in turn is 1000 times faster than BEAST on their simulated dataset of 446 taxa – yet it performs as well as or better than popular Bayesian methods. It also outperforms Bayesian methods when evolutionary rates vary drastically across the tree. One exciting aspect of the method is that, according to the authors, “the branch (relative) rates produced by RelTime directly reveal the statistical properties of the distribution of evolutionary rates in a phylogeny, which exposes clades and lineages with significantly slower or faster evolutionary rates.” For a lab studying the radiation of the Hawaiian Drosophila, this is an important application. There are multiple examples in the clade where evolutionary rates probably exploded when a lineage colonized a new island or niche. RelTime looks like it is worth trying as a way to rapidly assess this phenomenon. Brian Attempting to estimate divergence dates on molecular phylogenies is a messy affair. Bayesian methods (for example, BEAST and MrBayes) have the advantage of being able to accommodate a lot of unknowns and still often are able to generate distributions that seem to contain the true likely divergence date of interest. Three classes of information can be included in a Bayesian analysis of divergence dating to calibrate the phylogeny: molecular rates, biogeographic information, and fossils. Fossils tend to be thought of as the gold standard among these three – researchers take the date for the oldest known fossil from their group of interest and apply it as a calibration on a node in their phylogeny. However, this presents a few issues. First, fossils are often imperfectly preserved, making them difficult to place taxonomically. Second, it can be difficult to come up with appropriate prior distributions to apply in the dating machinery. In addition, in current dating methods, researchers have to assume that they know for certain on which group’s node to place a fossil calibration and that the group is monophyletic – both of these can be strong assumptions.
In a new paper by Ronquist et al (2012), researchers present a new approach for dealing with these issues, using MrBayes 3.2. Instead of placing a calibration on a node in the phylogeny, the fossil is included as a species in the data matrix. How it works is that extant species in the data set have molecular and morphological characters included, while the extinct species are coded for all the morphological characters that can be assigned (usually a subset of all of the characters because it is often not possible to see all of the characters on a fossil). Rather than calibrating nodes of groups the user defines, the user can enter the dates of the strata the fossils came from as a calibration on the taxa itself. The Mr.Bayes machinery then takes this information and estimates a phylogenetic tree, placing the fossils alongside the extant taxa based on their morphological characters and using their dates to estimate divergence times for the rest of the tree. This approach is neat because it removes error associated with improperly classifying fossils and having to restrict the node it is placed on to be monophyletic. Ronquist et al (2012) test the method on a data set of the early Hymenoptera radiation that includes 45 fossils, many of which are poorly preserved, and also run a comparative analysis using the more traditional node-dating technique. One issue they had to deal with in the development of this approach was that they did not feel that existing tree priors (e.g. birth-death, Yule) could be reasonably applied to their data set. So, they developed and described a new, uninformative tree prior that allows the tree to have terminals of different ages, which allows the branch length information to come from the data. Because this was a proof-of-concept paper, they did extensive exploration of the method, sensitivity analyses, and took a lot of care with selecting their prior distributions. They first ran an uncalibrated analysis of the data, from which they could detect significant rate variation among the lineages. Because of this, they did not want to use a strict clock for the analysis (which assumes rates are the same along all of the branches in the tree). Instead, they wanted to allow for the rates to vary among the branches, so used a relaxed clock approach. There are several relaxed clock models available that model how rates vary in very different ways. As it was not clear which model fit their data best, they ran the analysis with three different models (two autocorrelated models and one uncorrelated model) and compared them using Bayes Factors. In addition to helping them identify the best model for their data, this allowed them to showcase one of the many new features in MrBayes 3.2, calculation of the marginal likelihoods using stepping-stone sampling. This is a major advance for MrBayes users, allowing them to do model comparisons and hypothesis testing without having to use the harmonic mean estimator of the likelihood, which is known to produce biased estimates. Plus, it was a really nice description for users of a method for going about accounting for rate variation for their own data sets. In their final comparison of the Total-Evidence approach versus the node dating approach, Ronquist et al found several interesting things. First, they found that their method produced a tree that compared well topologically and in its estimates of divergence times with previous studies. They also found that their method is both more precise (smaller error bars) and less sensitive to prior choice than the node dating approach. Finally, the Total-Evidence analysis produced posterior probability distributions of less than 50% for over half of the fossils used as node calibrations in the node dating approach (even though the authors had sub-selected only the best-understood fossils in the group), indicating that their placement in the tree is highly uncertain, and thus indicating that specifying which node they belong to is inappropriate. Ronquist et al’s findings suggest that this is a method worth continuing to explore. Carefully selecting models and priors and running sensitivity analyses will be important for users as they begin testing this on new data sets. The authors provide their nexus file with all of the blocks of commands as supplementary information, so you can see for yourself how these elegant analyses are set up. Kari Goodman Ronquist, F., Kloppstein, S., Vilhelmsen, L, Schulmeister, S., Murray, D.L. and A.P. Rasnitsyn 2012. A Total-Evidence Approach to Dating with Fossils, Applied to the Early Radiation of the Hymenoptera. Systematic Biology 61 (6): 973-999. 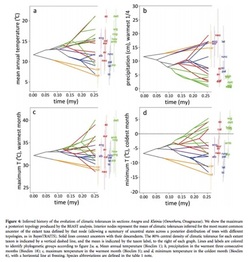 The other day I got into a class discussion about ancestral state reconstruction, or the assignment of ancestral states based on extant taxa. Some people thought that character mapping was not scientifically sound unless you had a time machine. I, however, disagree. The utility of phylogenetics is to determine evolutionary relationships between extant taxa, and while time machines do not exist, systematists are able to make hypotheses about these relationships. I think character mapping follows the same principles and gives science more insight into diversification processes. However, one problem I do see with ancestral state reconstruction is that your analysis is only as good as your phylogenetic tree. As many people say, “junk in, junk out.” Therefore, use with caution.
One new ancestral state reconstruction method I recently learned uses the software package Phyloclim maintained by Christoph Heibl. Phyloclim is a package implemented in R that integrates species ecological niche models with phylogenetics in order to calculate and visualize niche evolution on a phylogenetic tree. To do this, the user must first create a predicted species occupancy model. This involves using ecological niche modeling software, such as Maxent, and species location data stored as a raster file--I believe this is done in a mapping program like GIS. There is a great tutorial on the Maxent website. Patterns of niche evolution or niche conservation can then be determined by comparing the variation between niche space within and between subclades. One of two outcomes is expected: 1) either niche evolution occurs between subclades with conservation of niche space within each subclade or 2) niche conservation occurs between subclades and niche evolution occurs within each subclade. This means you could either see a divergence of niche spaces at the beginning family followed by conservation of their niche spaces or you would see niche space preserved early on in evolutionary history followed by diversification among species. Inference can then be made about ancestral niche spaces using a statistical analysis, and a statement of how climate (or other variables) may or may not result in the diversification of a group. I really liked using this R package. The visual of ancestral state reconstruction (see figure above) is really informative and neat to look at. However, Phyloclim is still quite new and no formal tutorial from the developers exists, but there are examples in the software package complete with example data. I have yet to try using my own data in Phyloclim, but I think it’s as simple reading a .csv file of your ecological niches models and your tree file in newick or nexus format (I like this tutorial) into R. I realize this may be easier said then done, but in theory it’s simple. Jessica Craft Useful papers: Evans, M. E. K., S. A. Smith, R. S. Flynn, and M. J. Donoghue. 2009. Climate, niche evolution, anddiversification of the ’bird-cage evening primroses’ (Oenothera, sections Anogra and Kleinia). Am. Nat. 173: 225-240. Fitzpatrick, B.M & Turelli, M. 2006. The geography of mammalian speciation:mixed signals from phylogenies and range maps. Evolution 60: 601-615. Phillips, S.J, M. Dudik, & R.E. Schapire. 2006. Maximum entropy modeling of species geographic distributions. Ecological Modeling 190: 231-259. Warren, D., R.E. Glor, & M. Turelli. 2008. Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62: 2868-2883 B
The paper “Climate, Niche Evolution, and Diversification of the “Bird‐Cage” Evening Primroses (Oenothera, Sections Anogra and Kleinia)” from the journal The American Naturalist (2009) uses the relatively new technique of combining niche models and dated phylogenies to make conclusions about the drivers of biodiversity. Integrating ecological niche modeling into phylogenetics is a potentially powerful practice that could help us answer questions about the creation and sustaining of biodiversity. These questions are becoming increasingly important with large-scale anthropogenic disturbances to the environment including habitat destruction, landscape fragmentation and, of course, climate change threaten biodiversity globally. While, “the use of niche models to address paleocli- matic explanations for diversification has only begun,” this paper seems to be one of many studies on the topic, and the jury is still out on what the actual drivers of biodiversity are, as the results of the papers are clearly at odds with each other. This paper does a good job of citing previous studies that have gotten results that are both similar and opposing. However, for the reader, it lessens the credibility of the study itself. Why is this study any different or more believable than the others? This study seeks to fragment the question by concluding that different clades are driven in different ways. Furthermore, it concludes that the study itself offers a new method upon which other studies can build: “ Here we have provided a method, beginning with climate and locality data, via ecological niche modeling, for reconstructing ancestral climatic tolerances that takes into account both the intraspecific variability of extant taxa and phylogenetic uncertainty.” Integrating phylogenies and niche data to infer information about the drivers of diversity seems like a good idea that can help us answer essential questions. While this study seeks to find some answers to these big questions, it proves to be just another building block in our understanding and application of this new process. Perhaps the methods laid out in this paper will be useful in future studies to come up with more definitive answers, or perhaps they will add further support to differing findings on the subject. I guess that is the nature of what we do. We set up studies to answer big important questions that, slowly (this is ideally speaking) can gain momentum until someone can place a capstone on top and society embraces a new theory. I just hope that we as a species can understand the drivers of biodiversity before we forever deplete it. We're scheduled to host happy hour on the 15th at 1130 in the new Mulford conference room. Please bring something to share with the department.
 Teri Markow and I have a chapter comparing patterns of diversification in Hawaiian Drosophila and the cactophilic repleta species group.
|
PatrickProfessor Archives
April 2018
Categories
All
|
 RSS Feed
RSS Feed
