
The 11th Workshop on Drosophila Species Identification and Use was held in San Diego from 17-21 October.
| O'Grady Lab |
|
|
The 11th Workshop on Drosophila Species Identification and Use was held in San Diego from 17-21 October.
0 Comments
As a fly enthusiast, I understand how daunting a task identifying species can be. The minute details, the crazy terms: it can all make you lose your head, especially when you’ve gathered a seemingly infinite amount of specimens. But, what’s a scientist to do?
You could hunker down at a microscope and wait until your eyes cross, or you could head down the road of genetic barcoding. Now, simmer down, you taxonomists. I don’t plan to argue you guys out of your jobs. In fact, I have my own criticisms of barcoding, but just humor me for a moment. Genetic barcoding works by sequencing small DNA portions from unknown organisms and comparing those sequences to a barcode library. So say you’ve collected a bunch of something, let’s say unicorns from the North Pole as everyone knows all magical ponies live in the wintery north. Well, as a well-known unicorn scientist you are aware that there are several cryptic species of unicorns. This means that two or more species appear morphologically similar but, by at least one of the many species concepts, are still considered separate species. A quick PCR analysis, PCR gods forgiving, and a BLAST to the NCBI database could tell you which mythical unicorn species you now possess (should the barcode library of unicorns be complete). Okay, I may have lied. Unicorns don’t really exist (outside the imagination of yours truly), but the problem of cryptic species does, along with a myriad of other identification issues such as morphological variation within species and even between adults and juveniles. Have you ever looked at drosophila larvae? They all look like squiggly, little, wormy things, every single one of them. Aside from some neat distinguishing behaviors – a few fling themselves like trapeze artists – you couldn’t tell them apart. So, it makes sense that a useful tool like barcoding has received so much attention, but let’s not get carried away. This isn’t the messiah come here to solve all our problems. The way I see it genetic barcoding is the microwave of the 1970’s housewife: a new tool for the modern taxonomist. It heats your food in mere minutes, but you can still burn the pot roast. Criticisms include incorrectly identified species sequences, a substantial error rate, and lowered ability to distinguish between recently diverged species. These comments all point towards the necessity of well-studied taxonomists to make final decisions. Me? I’m sticking to the microscope for now. Having a good grasp on taxonomic identification seems like it will always be a useful tool. Jessica Craft Not many people get paid to be twelve years old, at least not as adults, so I feel I’m one of the lucky ones. I’ve been working on a project that lets me go to so some beautiful rivers and streams, flip over rocks, and look for aquatic insects. It kindles the fun and curiosity that I remember while doing that kind of thing when I was a kid. Now, of course, I have a research question in mind while I’m out there. Our lab has been conducting surveys of aquatic insects in a few representative Northern California watersheds to establish the composition of aquatic insect communities, create a DNA barcoding (see this blog, too) database of Norcal aquatics for more efficient biomonitoring in the future, link taxa to characteristics of the habitat, and, using landscape genetics, make predictions about how global change biology may affect our local rivers and streams.
Aquatic insects have been used in biomonitoring for about a century as a way to assess the health of riparian areas. Biomonitoring adds informative data to chemical testing of water. Chemical testing provides valuable information about a particular component, such as dissolved oxygen or the concentration of a pollutant, at one moment in time. Biomonitoring is a way to assess whether all of the components of a system are such that they support the surveyed organisms over their entire lifespan. Both chemical and biological surveys can be combined to give a fuller picture of ecosystem health. Biomonitoring of aquatic insects is now being used not only to assess current and past ecosystem health, but also to predict future changes, for example in response to climate change. In recent years, concerns about the effects of human-driven climate change on riparian ecosystem have increased. Climate change is projected to alter precipitation patterns, the timing of seasonal transitions, and extremes of both heat and cold, among other effects. These changes will affect different members of biotic communities differently according to their ability to adapt to changing conditions or disperse to more favorable habitat. We can use species distribution modeling to identify key characteristics of favorable habitat, and use patterns we find today using landscape genetics to identify potential obstacles that could prevent taxa from shifting ranges. We are fortunate to be doing this as part of a larger consortium on campus, the Berkeley Initiative in Global Change Biology, or BIGCB. With funding from the Vice Chancellor’s Office, the Moore Foundation and the Keck Foundation, the BIGCB is focused on global change forecasting for California ecosystems, using analyses of fossil, historic and current data to better understand California ecosystems responses to environmental change and make predictions of future ecosystem changes. Brian Ort Which genomic changes underlie rapid adaptation? Do these adaptations come from new mutations or from genetic variation already existing in ancestral populations? Are the genomic regions found in protein coding or regulatory regions? This list of questions reads like the intro to a Trends in Ecology and Evolution article on hot questions in evolutionary biology, and is what Jones et al (including David Kingsley) approached in 7 pages of awesome, detailed work on the genome biology of sticklbacks.
Threespine sticklebacks are famous in the evolution world as a study system for rapid adaptation and speciation. In separate populations all over the world, they invaded from the ocean to adapt to the new freshwater environments created after the Pleistocene glaciers retreated about 11,000 years ago (evolutionarily speaking, this was very recent). This created naturally replicated marine/freshwater population pairs that still hybridize in nature. They even can be raised in a lab, which means they can also be experimented on. We know that all plants and animals have adapted to changing conditions at some point or another in their history, but the process is difficult to study in many organisms, and often the genomic signatures of such change are often obscured by the effects of too much time having passed. The sticklebacks have a perfect storm of attributes that make them great for studying these sorts of questions. In a paper that begins by presenting the first threespine stickleback genome (which is exactly as far as many first genome papers go), Jones and collegues then go deeper into the system, using that genome to look in detail at how it responded to such recent and drastic environmental change. They leverage the power of the naturally replicated freshwater invasions by generating 20 additional genomes from marine/freshwater population pairs all over the world. In order to assess parallel changes occurring across the entire genome, they looked for regions in the genomes that were similar among all the freshwater animals worldwide but different from the marine ones. Using two complimentary approaches, they found 147 regions (0.2% of the whole genome) that were divergent among the ecotypes. The researchers then focused in on one marine/freshwater population pair with an active hybrid zone to ask if these globally shared variants were the main ones involved or if there were also a lot of variants contributed by the local populations. They found of the divergent changes between the two populations, 35.3% contained these global variants, suggesting that there is a substantial contribution from local variants in each population in addition to what is shared across populations. An outstanding question in biology asks whether adaptive changes occur because of changes in protein coding genes or regulatory regions. Evidence has been accumulating from a variety of systems about specific adaptations, which are typically restricted to relatively narrow regions within a genome. This study allowed a look at what is going on across an entire genome. The authors found that of all of the freshwater variants that were shared across all populations, 17% were located within protein coding regions, while 41% were found in non-coding regions and presumed to be regulatory. An additional 43% were more ambiguous, and the authors speculate that they also primarily fall into the regulatory category. More work needs to be done to classify and verify these variants, but the results are already suggestive that a significant amount of adaptive change across the genome is due to changes in the regulatory regions. While we are not quite at a stage of being able to write a how-to manual on adapting to a novel environment, this series of studies provides a lot of new detail on how it works in nature in one particularly well-suited system – a system truly powerful and special for its ability to give us insight into the dynamics of rapid adaptation. Even though the et al on this paper is a long list of contributors that render this approach way beyond what is possible to do by a single researcher, it is still inspirational to picture how these approaches could illuminate the biology of other natural systems. Kari Goodman Jones, F.C. et al 2012. The genomic basis of adaptive evolution in threespine sticklebacks, Nature, vol 484, pp. 55-61. doi:10.1038/nature10944. When I was 19 years old I visited the Organization of Tropical Studies’ La Selva Biological Station in Costa Rica. Upon a nature hike with a resident researcher, a hypothetical, nearly sci-fi idea was thrown out for ways to significantly improve field work. The scientist painted the picture of a futuristic pocket-sized chip that could puncture leaf or animal tissue, do a lightning fast DNA extraction and PCR, query a genetic database, and within minutes identify a specimen – right in the field! He proclaimed that this invention would allow scientists to categorize greater biodiversity, understand ecosystems more fully, and help to clarify the taxonomy and phylogeny of tropical species.
Daniel Janzen, a renowned tropical ecologist and professor at the University of Pennsylvania, is a major proponent of this theoretical device. Janzen has been involved in the 'Consortium for the Barcode of Life’ project, which includes members such as the Natural History Museum in London, the Smithsonian in the US, the University of Guelph in Canada, Rockefeller University in New York, and a host of other institutions. The goal of this research consortium is to use a single DNA sequence, (cytochrome oxidase I, a mitochondrial gene), to essentially tag, or “barcorde” every species on earth. Having one gene with which to identify all biodiversity is a lofty task that will require many skilled technicians in functioning genetic labs, as well as taxonomic experts to assign appropriate names and voucher specimens to all of these sequences. Still Janzen suggests that with the use of the proposed ‘gene chip’ the process could be conducted by a “six-year kid walking down the street.” Progress has already been made in the construction and usage of this 'theoretical' device. Mesa Tech International has developed the ‘DNA dipstick,’ a hand-held, battery-powered, disposable device that can identify nucleic acid sequence-level data within hours. This device has been used to identify microbial pathogens in agricultural crops and animals and thus improve human health. DNA microarrays have also been used in the Fish&Chips project which hopes to identify and categorize marine biodiversity. This project uses a ‘bio-chip’ made of glass that contains oligionucleotides fixed to the chips’ surface, which acts as a probe to bind complementary target DNA sequences by hybridization. This group also has a Phytoplankton Chip and Invertebrate Chip. With such technological developments in recent years, the quick identification of specimens in the field, as proposed by the Costa Rican researcher some years ago, suggests that this goal is not so far-fetched. DNA barcoding and the use of gene-chips will undoubtedly herald science into a new era, as we begin to database and identify genes of all of earth’s species. Iman Sylvain Several recent events have me reflecting on how science is done and how different schools of scientific thought turn over through time. I'm teaching a grad class in phylogenetic methods for the first time since 2007 and I've noticed a big difference in the students. In the past, their emphasis has been on understanding the nuts and bolts of how to generate phylogenies. While the students this year are still interested in building trees, I'm getting the sense that they view tree building as a means to an end, rather than a valid activity in and of itself. Student interest seems to have shifted more towards using trees to test evolutionary hypotheses. While the sample size is small, this echoes what I've heard from colleagues about two courses taught in integrative biology, IB200A (phylogenetic reconstruction) and IB200B (phylogenetic hypothesis testing). Enrollment in 200A is dropping relative to 200B.
It's an interesting phenomenon and makes me think we may be in the midst of another paradigm shift (albeit a small one) in how systematics is done. Looking back over the years, you can clearly see turnovers in schools of scientific inquiry. Here's a short list: Numerical Taxonomy Starting in the early 1960s, numerical taxonomists brought a quantitative approach to taxonomy and systematics that previously been absent. This was driven largely by statisticians (Sokal, Sneath) and the notion that careful measurements could lead to improved taxonomic hierarchies. During the 1970s this field fractured into phenetics and cladistics. Then the cladists ate all the pheneticists. Cladistics (and cladists) While cladistics and cladists are tightly linked, not all people who practice cladistics are cladists and not all cladists always employ a strictly cladistic approach. In essence, it's a semantic argument, something that all good cladists enjoy. I define cladists as those followers of Willi Hennig who espouse a parsimony-only approach to systematics. They aggressively routed the numerical taxonomists in the 70s and then stuck around to rail against likelihood, Bayesian analysis, and, in some cases, evolutionary inference itself. For more detail on this era of systematics, check out the chapter in Joe Felsenstein's Inferring Phylogenies book. Molecules vs. morphology Starting in the mid-1980s, the introduction of PCR led to a technical revolution in systematics. Suddenly, everyone was scrambling to sequence DNA in his or her favorite organism and use it to generate phylogenies. Like most of the previous theoretical and technical advances in systematics, DNA promised to "fix everything." This, of course, hasn't come to pass and, even now that we can sequence entire genomes, some systematic questions remain difficult to approach. What did happen was a massive shift in resources, both in terms of grant funding and jobs offered, with the traditional morphologists being on the losing end of things. This led to a lot of animosity - I can still remember being called a "moleculoid" by some of my older colleagues. Luckily, this has largely blown over and most systematists take a holistic approach to understanding relationships in their focal taxa. DNA barcoding In some ways barcoding is a spin off of the molecules vs. morphology debate. The notion here is that taxonomy isn't really needed now that we can use DNA sequence to uniquely identify (or barcode) species. While DNA approaches are important techniques to have in your taxonomic toolkit, throwing out all by a single character system (the COI gene if you work on animals) in your taxonomy is ridiculous. And many people have pointed this out before. The initial DNA barcoding push was really more of a marketing campaign than a novel scientific approach and, once again, a more inclusive approach is being taken. Statistical phylogenetics The idea that phylogenies are statistical statements about evolutionary history and can not only be viewed as hypotheses but also used to test hypotheses is the predominant paradigm in modern systematics. More advanced analytical techniques, increased processor speed, and the introduction of model-based approaches have all helped shaped modern phylogeneic systematics. Powerful statistical methods are currently causing an expansion of systematics and driving the "use of trees" over the "building of trees." I think this is a normal, natural outgrowth of the field and will hopefully continue to drive it forward. My own work is moving away from tree building and more into the area of community assembly and interaction so I've been reading a lot about phylogenetic community ecology (PCE from here on out - too much to write) as a way to merge the fields. I ran across this interesting blog post a few days back where the author, Jeremy Fox, makes the case that PCE is a "bandwagon." He makes bring up some good points (although he uses a pseudo-subjective literature review to do so) and the post is worth a read. This all leaves me wondering, however, if there's really anything wrong with any one field or subfield jumping on a bandwagon. This is, at least if you take an historical perspective, how science moves forward. For example, organismal biology jumped hard on the DNA bandwagon in the late 1980s/early 1990s, eliminating entire -ologies in the rush to capitalize on the new technology. Within 10 years, however, people began to realize that you couldn't place those DNA-based phylogenies in context without some knowledge of basic biology so the field corrected itself, including the new theories and technologies. I imagine this is what will happen as a result of the current push for hypothesis testing in phylogenetics. Patrick O'Grady 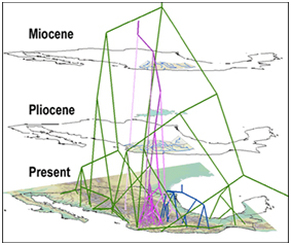 When we want to visualize biogeographical distributions we usually create maps. When we want to visualize phylogenetics we often build taxonomic trees. What if we want to visualize phylogeography? Typically we use maps and phylogenetic trees side-by-side. There is a relatively new tool called GeoPhyloBuilder that joins the two. It is available in ArcGIS 9.3 and later versions and was created by David Kidd and Xianhua Liu of The National Evolutionary Synthesis Center (2008). GeoPhyloBuilder builds a 3D spatiotemporal, phylogenetic GIS data model by attaching the phylogenetic tree tips to the geographical locations of the samples. The geographical locations can be points, lines, or polygons. The 3D dimension comes from the node depths of the phylogenetic tree. Longer, older branches are elevated further above the map. The model can be visualized in 2D or 3D in ArcMap, ArcScene, or other Earth Browsers. Examples of images and movies as well as the download are available at: https://www.nescent.org/sites/evoviz/GeoPhyloBuilder. Although some of these images make the phylogenetic tree look like spaghetti hanging over a map, you can color code different branches to see how they relate geographically. You can also visualize the 3D images in a movie, rotating the image so that you can get varying perspectives. Passing information on is easiest when you have powerful visuals and this may be helpful for some phylogeographical results.
Lisa Marrack Phylogenies of the freshwater fish family Goodienae: (purple; Webb et al., 2004) and genera Poeciliopsis (green; Mateos et al., 2002) and Notropis (blue; Schonhuth & Doadrio, 2003) with modern elevation and drainage. Pliocene and Miocene drainage and palaeolakes from de Cserna & Alvarez (1995). [In Kidd and Ritchie (2006): Journal of Biogeography]. Review: Webb et al. (2011) DNA Barcoding of North American Ephemeroptera: Progress and Prospects10/12/2012  This paper was a convincing argument for the promise of DNA barcoding taking over the world, basically. DNA barcoding of aquatic macroinvertebrates is gaining backing as an extremely useful tool for taxonomic identification and research, and in turn, application in bioassessment programs. Some have argued that DNA barcoding is an unreliable way to identify aquatic macroinvertebrates, but this paper shoots those ideas down; (!!!) as it found that the average intraspecific divergence was 12.5%, while the average intraspecific divergence was 1.97%. While there were some complications in identification, caused mainly by polyphyly and species complexes (which still need to be further studied and resolved,) in general these results indicate that DNA barcoding is, in general, a promising tool in aquatic macroinvertebrate taxonomy and bioassessment programs.
Aside from the intra and interspecific divergences being accurate, for the most part, this paper further points out that DNA barcoding is particularly useful for other reasons. In addition to helping streamline the identification, delimitation, and discovery of species, DNA barcoding also gives consistent results across life stages, which is particularly important in aquatic ecology applications, as a large majority of benthic macroinvertebrates are immature. In many cases, taxonomy is based on adult male morphology, and identification of immatures, particularly early instars, is exceedingly time-consuming and requires substantial training. Additionally, specimens are often very tiny, and delicate, which can lead, in many cases, to missing gills, caudal filaments or even legs, which can in turn further complicate accurate identifications. Furthermore, the use of DNA barcoding allows for data standardization, and thus a broader, more accurate comparison of results. This paper also suggested that much more work on North American Ephemeroptera taxonomy and classification is required, as many currently recognized species are highly divergent. Most of these confused species have complex histories of synonymy and reflect the 60 year trend in North American mayfly systematics towards inclusive species concepts. Further taxonomic work that synthesizes a variety of identification and classification methods including morphological, biogeographic, ecological, behavioral and molecular techniques is required to test current species hypotheses, particularly of those unusually divergent Ephemeroptera species. DNA barcoding is one of the techniques that will be useful in this aim of achieving stable, supported species hypotheses. Re-examined and updated species hypotheses will allow us to identify aquatic insects more accurately and more efficiently, which will in turn allow us to determine and communicate the ecological characteristics of a species, such as phenology and tolerance to pollutants, and thus improve our ability to utilize these organisms in bioassessment programs. Natalie Stauffer |
PatrickProfessor Archives
April 2018
Categories
All
|
 RSS Feed
RSS Feed
